Koselugo has a consistent dosing schedule that helps patients and caregivers maintain their routine
Recommended dosage
25 mg/m2 | twice daily
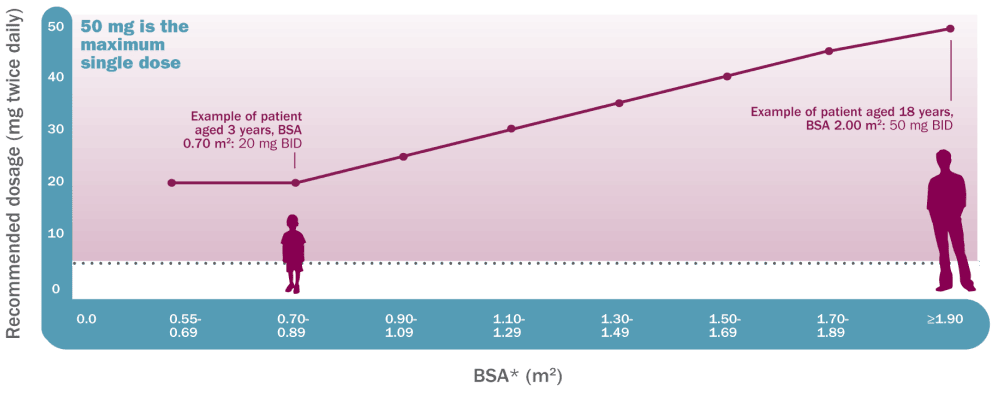
Dosing is individualized based on BSA (mg/ m2) and is rounded to the nearest achievable 5-mg or 10-mg dose (up to a maximum single dose of 50 mg).

It is important to monitor your patients and reassess dosage based on their BSA changes.
Koselugo is available in 25-mg and 10-mg capsules and 28- and 60-count bottles

Recommended administration of Koselugo:
- Before prescribing, children should be assessed for the ability to swallow capsules
- Orally twice daily (approximately every 12 hours) until disease progression
-
No fasting requirement

- Swallowed whole with water: Do not chew, dissolve, or open capsule
Advise patients:
Do not take a missed dose unless it is more than 6 hours until the next scheduled dose
If vomiting occurs, do not take an additional dose but continue with the next scheduled dose
Testing requirements1
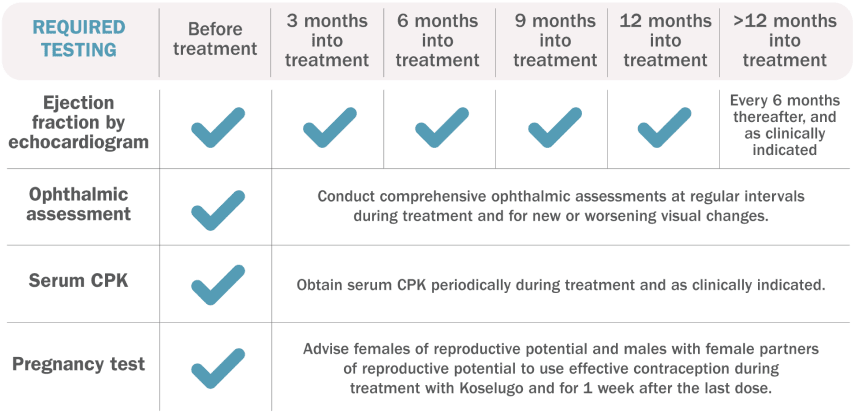
Additional Evaluation Guidelines1:
Ejection Fraction by Echocardiogram: Withhold, reduce dose, or permanently discontinue Koselugo based on severity of adverse reaction. In patients who interrupt Koselugo for decreased LVEF, obtain an echocardiogram or a cardiac MRI every 3 to 6 weeks. Upon resolution of decreased LVEF to greater than or equal to the institutional LLN, obtain an echocardiogram or a cardiac MRI every 2 to 3 months or as directed by the cardiologist.
Ophthalmic Assessment: Permanently discontinue Koselugo in patients with RVO. Withhold Koselugo in patients with RPED, follow up with optical coherence tomography assessments every 3 weeks until resolution, and resume Koselugo at a reduced dose. For other ocular toxicities, withhold, reduce dose, or permanently discontinue Koselugo based on severity of adverse reaction.
Serum CPK: If increased CPK occurs, evaluate patients for rhabdomyolysis or other causes. Withhold, reduce dose, or permanently discontinue Koselugo based on severity of adverse reaction.
Pregnancy Test: Assess the pregnancy status of females of reproductive age. Advise pregnant women of the potential risk to a fetus.
BSA=body surface area; CPK=creatine phosphokinase; LLN=lower limit of normal; LVEF=left ventricular ejection fraction; MRI=magnetic resonance imaging; RPED=retinal pigment epithelial detachment; RVO=retinal vein occlusion.
Koselugo dosing is based on BSA1
Recommended dosage based on BSA
Recommended dosage and dose reductions for adverse reactions on Koselugo
| BSA |
Initial Koselugo Dose*
(mg/twice daily)
|
First dose reduction
(mg/dose)
|
Second dose reduction†
(mg/dose)
|
|---|---|---|---|
|
0.55-0.69 m2
|
20
10
|
10
10
|
10 mg once daily
|
|
0.70-0.89 m2
|
20
20
|
20
10
|
10
10
|
|
0.90-1.09 m2
|
25
25
|
25
10
|
10
10
|
|
1.10-1.29 m2
|
30
30
|
25
20
|
20
10
|
|
1.30-1.49 m2
|
35
35
|
25
25
|
25
10
|
|
1.50-1.69 m2
|
40
40
|
30
30
|
25
20
|
|
1.70-1.89 m2
|
45
45
|
35
30
|
25
20
|
|
≥1.90 m2
|
50
50
|
35
35
|
25
25
|
*The recommended dosage for patients with a BSA less than 0.55 m2 has not been established.
†Permanently discontinue Koselugo in patients unable to tolerate Koselugo after 2 dose reductions.
BID=twice daily; BSA=body surface area.
Hepatic Impairment‡
Drug Interactions‡
‡For recommended dosage in hepatic impairment and dosage modifications due to drug interactions, see Tables 4 and 5 in the Prescribing Information.
Dosage modifications1
Recommended dosage modifications for adverse reactions on Koselugo§
Severity of adverse reaction
Recommended dosage modifications for Koselugo
Cardiomyopathy
- Asymptomatic decrease in LVEF of 10% or greater from baseline
and less than LLN
Withhold until resolution.
Resume at reduced dose.
- Symptomatic decreased LVEF
- Grade 3 or 4 decreased LVEF
Permanently discontinue.
Ocular toxicity
- RPED
Withhold until resolution. Resume at reduced dose.
- RVO
Permanently discontinue.
Gastrointestinal toxicity
- Grade 3 diarrhea
Withhold until improved to Grade 0 or 1.
Resume at same dose.
Permanently discontinue if no improvement within 3 days.
- Grade 4 diarrhea
Permanently discontinue.
- Grade 3 or 4 colitis
Permanently discontinue.
Skin toxicity
- Grade 3 or 4
Withhold until improvement.
Resume at reduced dose.
Increased CPK
- Grade 4 increased CPK
- Any increased
CPK and myalgia
Withhold until improved to Grade 0 or 1.
Resume at reduced dose. Permanently
discontinue if no improvement within 3 weeks.
- Rhabdomyolysis
Permanently discontinue.
Other adverse reactions
- Intolerable Grade 2
- Grade 3
Withhold until improved to Grade 0 or 1.
Resume at reduced dose.
- Grade 4
Withhold until improved to Grade 0 or 1.
Resume at reduced dose. Consider discontinuation.
§Per National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Cardiomyopathy. A decrease in left
ventricular ejection fraction (LVEF) ≥10% below baseline occurred
in pediatric
patients who received Koselugo in SPRINT with some
experiencing decreased LVEF below the institutional lower limit of normal
(LLN), including one patient with Grade 3. All patients with decreased
LVEF were asymptomatic and identified during routine echocardiography.
The safety of Koselugo has not been established in patients with a history
of impaired LVEF or a baseline ejection fraction that is below the institutional
LLN. Assess ejection fraction by echocardiogram prior to initiating treatment,
every 3 months during the first year of treatment, every 6 months thereafter,
and as clinically indicated. Withhold, reduce dose, or permanently discontinue
Koselugo based on severity of adverse reaction. In patients who interrupt
Koselugo for decreased LVEF, obtain an echocardiogram or a cardiac MRI
every 3 to 6 weeks. Upon resolution of decreased LVEF, obtain an echocardiogram
or a cardiac MRI every 2 to 3 months.
Ocular Toxicity. Blurred vision,
photophobia, cataracts, and ocular hypertension
occurred. Retinal
pigment epithelial detachment (RPED) occurred in the pediatric population
during treatment with single agent Koselugo and resulted in permanent discontinuation.
Conduct ophthalmic assessments prior to initiating Koselugo, at regular
intervals during treatment, and for new or worsening visual changes. Permanently
discontinue Koselugo in patients with retinal vein occlusion (RVO). Withhold
Koselugo in patients with RPED, conduct ophthalmic assessments every 3
weeks until resolution, and resume Koselugo at a reduced dose.
Gastrointestinal Toxicity. Diarrhea
occurred, including Grade 3. Diarrhea resulting in permanent discontinuation,
dose interruption or dose reduction occurred. Advise patients to start
an anti-diarrheal agent (eg, loperamide) and to increase fluid intake immediately
after the first episode of diarrhea. Withhold, reduce dose, or permanently
discontinue Koselugo based on severity of adverse reaction.
Skin Toxicity. Rash occurred in 91% of 74 pediatric patients. The most frequent rashes included dermatitis acneiform (54%), maculopapular rash (39%), and eczema (28%). Grade 3 rash occurred, in addition to rash resulting in dose interruption or dose reduction. Monitor for severe skin rashes. Withhold, reduce dose, or permanently discontinue Koselugo based on severity of adverse reaction.
Increased Creatine Phosphokinase (CPK). Increased CPK occurred, including Grade 3 or 4 resulting in dose reduction. Increased CPK concurrent with myalgia occurred, including one patient who permanently discontinued Koselugo for myalgia. Obtain serum CPK prior to initiating Koselugo, periodically during treatment, and as clinically indicated. If increased CPK occurs, evaluate for rhabdomyolysis or other causes. Withhold, reduce dose, or permanently discontinue Koselugo based on severity of adverse reaction.
Increased Levels of Vitamin E and Risk of Bleeding. Koselugo capsules contain
vitamin E which can inhibit platelet
aggregation and antagonize vitamin K-dependent clotting factors. Supplemental
vitamin E is not recommended if daily vitamin E intake (including the amount
of vitamin E in Koselugo and supplement) will exceed the recommended or
safe limits due to increased risk of bleeding. An increased risk of bleeding
may occur in patients who are coadministered vitamin-K antagonists or anti-platelet
antagonists with Koselugo. Monitor for bleeding in these patients and increase
international normalized ratio (INR) in patients taking a vitamin-K antagonist.
Perform anticoagulant assessments more frequently and adjust the dose of
vitamin K antagonists or anti-platelet agents as appropriate.
Embryo-Fetal Toxicity. Based on
findings
from animal studies, Koselugo can cause fetal harm when
administered during pregnancy. In animal studies, administration of selumetinib
to mice during organogenesis caused reduced fetal weight, adverse structural
defects, and effects on embryo-fetal survival at approximate exposures
>5 times the human exposure at the clinical dose of 25 mg/m2 twice daily.
Advise patients of reproductive potential of the potential risk to a fetus
and to use effective contraception during treatment with Koselugo and for
1 week after the last dose.
ADVERSE REACTIONS
Common adverse reactions ≥40% include vomiting, rash (all), abdominal pain, diarrhea, nausea, dry skin, musculoskeletal pain, fatigue, pyrexia, acneiform rash, stomatitis, headache, paronychia, and pruritus.
DRUG INTERACTIONS
Effect of Other Drugs on Koselugo
Concomitant use of Koselugo with a strong or moderate CYP3A4 inducer decreased selumetinib plasma concentrations, which may reduce Koselugo efficacy. Avoid concomitant use with Koselugo.
SPECIAL POPULATIONS
Pregnancy & Lactation. Verify the
pregnancy status of patients of reproductive potential prior to
initiating Koselugo. Due to the potential for adverse reactions in a breastfed
child, advise patients not to breastfeed during treatment with Koselugo
and for 1 week after the last dose.
INDICATION
KOSELUGO is indicated for the treatment of pediatric patients 2 years of age and older with neurofibromatosis type 1 (NF1) who have symptomatic, inoperable plexiform neurofibromas (PN).
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca
1-800-236-9933
or at
https://us-aereporting.astrazeneca.com
or FDA at
1-800-FDA-1088 or
www.fda.gov/
medwatch.
Please see full Prescribing Information for Koselugo® (selumetinib).
References:
1. Koselugo. Package insert. AstraZeneca Pharmaceuticals LP.